Die Polymerase Chain Reaction
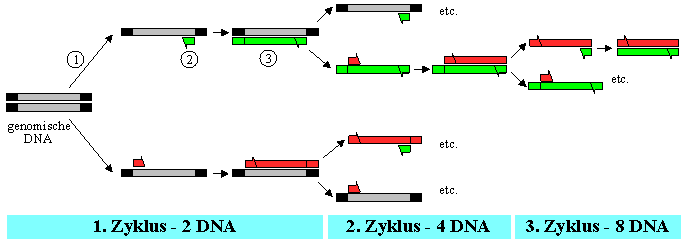
Die Polymerase Chain Reaction (PCR) ist eine universelle Methode milliardenfache Kopien spezifischer Fragmente von DNA’s in vitro zu synthetisieren. Dazu müssen lediglich die Start- und Endsequenzen des betreffenden Abschnittes bekannt sein und für jeweils komplementäre Stränge mit einer Größe von ca. 15-20 Nucleotiden synthetisiert werden. Diese Oligonucleotide bezeichnet man als "Primer".
Verantwortlich für die Synthese der neuen Stränge ist die DNA-Polymerase, die aus einem hoch-thermophilen Bakterium (Thermus aquaticus) gewonnen wurde und auch durch wiederholtes Erhitzen nicht denaturiert.
In jedem Reaktionszyklus kann daher als erster Schritt eine thermische Trennung der beiden Stränge der genomischen DNA-Doppelhelix durchgeführt werden (¬ ). Durch Anwesenheit eines großen Überschusses der Primer hybridisieren die Einzelstränge beim Abkühlen bevorzugt mit den komplementären Start-Oligonucleotiden ( ). Mit den im Reaktionsgemisch vorhandenen Desoxyribonucleosid-Triphosphaten kann nun die DNA-Polymerase die fehlenden komplementären Stränge vom Primer an selektiv synthetisieren (® ).
Nach einigen Reaktionszyklen liegt dann als vorwiegendes Produkt nur eine einzige Art von DNA-Fragment vor, dessen Länge dem Abstand zwischen den beiden ursprünglichen Primern entspricht. In der Regel werden 20 bis 30 Zyklen durchgeführt, wobei jeder Zyklus etwa 5 Minuten dauert.
Lagern sich die Primer auch an unerwünschte Stellen oder andere DNA’s an, so kann man eine zweite PCR-Runde durchführen, um die Spezifität bei analytischen Verfahren zu erhöhen (Nested PCR). Man verwendet dazu Primer, die innerhalb des ersten Zielstranges liegen, sodaß in dieser zweiten Runde nur mehr Sequenzen vermehrt werden, die komplementär zum zweiten Primerpaar sind. Es ist dann sehr unwahrscheinlich, daß unerwünschte DNA-Bereiche, die mit Hilfe der ersten Primer vervielfältigt wurden, Sequenzen enthalten, die sich mit dem zweiten Primerpaar verbinden.
Durchführung der PCR:
(http://hdklab.wustl.edu/lab_manual/pcr/pcr1.html)
Time required:
1. 1-2 Days
2. PCR reaction: 3-6 hours or overnight
3. Polyacrylamide gel electrophoresis using "Mighty-small II" gel apparatus: 2.5 hours
4. Ethidium bromide staining and photography: 45 minutes
Special reagents:
1. Synthetic oligonucleotide primer pair flanking the sequence to be amplified
2. 5X PCR Buffer (250 mM KCl, 50 mM Tris-HCl pH 8.3, 7.5 mM MgCl2)
3. Mixture of four dNTPS (dGTP, dATP, dTTP, dCTP) each at 2.5 mM (Ultrapure dNTP set, Pharmacia #27-2035-01). The dNTP mixture is made by adding equal volumes of a 10 mM solution of each of the four separate dNTPs together.
4. Taq DNA Polymerase (AmpliTaqTM, Perkin-Elmer/Cetus)
5. Light mineral oil
6. Acrylamide (electrophoresis grade)
7. N,N'-Methylenebisacrylamide (electrophoresis grade, Ultra-Pure/BRL, #5516UB)
8. Ammonium persulfate (Ultra-Pure/BRL, #5523UA)
9. TEMED (N,N,N'N' Tetramethylethylenediamine, Ultra-Pure/ BRL, #5524UB)
Special Equipment:
1. Mighty-small II SE-250 vertical gel electrophoresis unit (Hoefer)
2. Perkin-Elmer/Cetus Thermal Cycler
3. Sterile Thin-wall 0.5 ml Thermocycler microfuge tubes: (TC-5, Midwest Scientific)
Recommendations for choosing oligonucleotide primers :
The aim is to choose oligonucleotide primers complementary to relatively unique sequences flanking the segment to be amplified. Although more rigorous calculations and considerations can be employed to choose optimal primers, a few general guidelines will be given below to supply a good starting point.
Primers for PCR are generally 20-30 bp long and are chosen to be complementary to one strand (5' to 3') upstream and complementary to the opposite strand (5' to 3') downstream from the sequence to be amplified. The 5' ends of the primers define the ends of the amplified PCR product. Primers should ideally contain relatively balanced GC vs. AT content (e.g. 45-55% GC), and no long stretches of any one base. Caution should also be taken that the two primers of the primer pair do not contain complementary structures >2 bp to avoid "primer dimer" formation resulting from annealing of the two primers (especially at their 3' ends). The target sequence to be amplified is ideally 200-400 bp in length, with an upper limit probably around 3 kb.
Procedure for polymerase chain reaction:
The PCR reaction can be performed in volumes from 5 µl to 200 µl or more. The protocol below is similar to that used by the Center for Genetics in Medicine when screening the YAC library using a specific PCR assay and is carried out in a 5 µl reaction volume. This volume is recommended when the purpose of the experiment is diagnostic (to visualize whether or not a specific product is generated). A scaled up volume can be used if the PCR product will be recovered from the gel or used for sequencing. The 5 µl reaction is performed in a 0.5 ml eppendorf tube and covered by a drop of oil before placing in the thermal cycler.
The following components will make up one reaction (5 µl total volume), but a cocktail of everything except the DNA will be made first:
Cocktail for 10 reactions
1.0 µl 5X PCR Buffer 10 µl 5X PCR Buffer
0.4 µl dNTP mixture (each at 2.5 mM) 4 µl dNTPs
0.2 µl* Primer pair (each primer at 25 µM) 2 µl Primer pair
(The primer pair solution is 1:1 mixture of the
50 µM primer solutions)
0.1 µl Taq polymerase 1 µl Taq polymerase
2.3 µl ddH2O 23 µl ddH20
plus,
1.0 µl DNA (100 ng genomic template DNA or < 50 ng cloned template)
*The range of final primer pair concentrations in a normal reaction mix is 0.25 - 2.5 µM and 0.5 µM is sometimes ideal.
Because of the small volumes involved, it is convenient to make a cocktail of the first five ingredients for each primer pair to be used. For instance, if 8 PCR reactions are to be performed from 8 different genomic or cloned DNA templates using one primer pair, then a cocktail may be made (including a slight excess) for 10 reactions by mixing together each of the volumes above multiplied by 10. A 4 µl aliquot of the cocktail will then be added to the 1.0 µl of DNA in each tube.
Steps:
1. Plan your experiment before adding any reagents (#primer pairs to be used, number of DNA templates, etc.). After doing so, make the appropriate cocktail/s and ensure complete mixing by tapping the tube and quick spinning. (N.B. Caution should be used to avoidcontamination of reactions with even small amounts of DNA. In addition, care should be taken to avoid contamination of pipetmen with carryover amplification products from previous reactions)
2. Pipet 4.0 µl of the appropriate cocktail directly into the bottom of a sterile microeppendorf tube for each reaction. The tubes should be labeled by placing a round sticker on the cap to prevent smearing by oil in subsequent steps.
3. Add 1.0 µl of the DNA directly into the drop of cocktail in each tube and ensure adequate mixing. Quick spin to collect the reaction mixture in the bottom of the tube.
4. Overlay each reaction with one drop of light mineral oil using a pasteur pipet. The samples may be quick spun if necessary before placing in the Perkin Elmer/Cetus PCR machine.
5. Place a drop of mineral oil into each well in the thermal cycler temperature block to be used for the samples (this ensures rapid temperature equilibration during cycling)
6. Place the tightly capped tubes in the temperature block and make sure each is firmly seated by pressing on the tubes individually.
The PCR machine must now be programmed for the specific reaction conditions desired (See brief operating instructions for Perkin-Elmer PCR machine). Each cycle in the polymerase chain reaction involves three steps (denaturing, primer annealing, polymerization), and the products are amplified by performing many cycles one after the other with the help of the automated thermal cycler. Refer to the literature citations at the end of this protocol for detailed explanation of the reaction. The Taq polymerase is heat stable, and remains active despite the high denaturing temperature of each cycle. A representative set of reaction conditions for 25-35 cycles is:
I. Denature 93-94 degrees C 1.5 minutes
II. Anneal 50-65 degrees C 2 minutes
III. Polymerize 72 degrees C 2 minutes
Strategies for optimizing PCR reactions are at the end of the protocol.
7. After completion of the PCR reaction, remove the tubes from the temperature block and wipe the outside free of excess oil before placing in an eppendorf rack.
8. Add 2.0 µl of 5X Ficoll stop dye directly into the aqueous phase "bubble" at the bottom of each tube, and then add 100 µl of chloroform:isoamyl alcohol (24:1) to each tube, shake well, and spin briefly.
9. Carefully remove only the aqueous "bubble" with a P20 pipetman set to 7-8 µl by placing the pipet tip against the bubble and slowly drawing it in. Each sample should then be placed in a separate clean eppendorf tube before loading onto the polyacrylamide gel.
10.The reaction products are conveniently separated according to size by electrophoresis through a 10% polyacrylamide "Mighty-small II" gel at 110 V for 2-2.5 hours, and visualized after staining the gel with ethidium bromide.
References:
B. Alberts, B. Bray, J.Lewis, M.Raff, K.Roberts und J.D.Watson, "Molekularbilogie der Zelle", 3. Auflage, VCH Verlagsgesellschaft mbH, 1997